MBTPS1 Related Disorders Research Group
About MBTPS1-related disorders for physicians and scientists
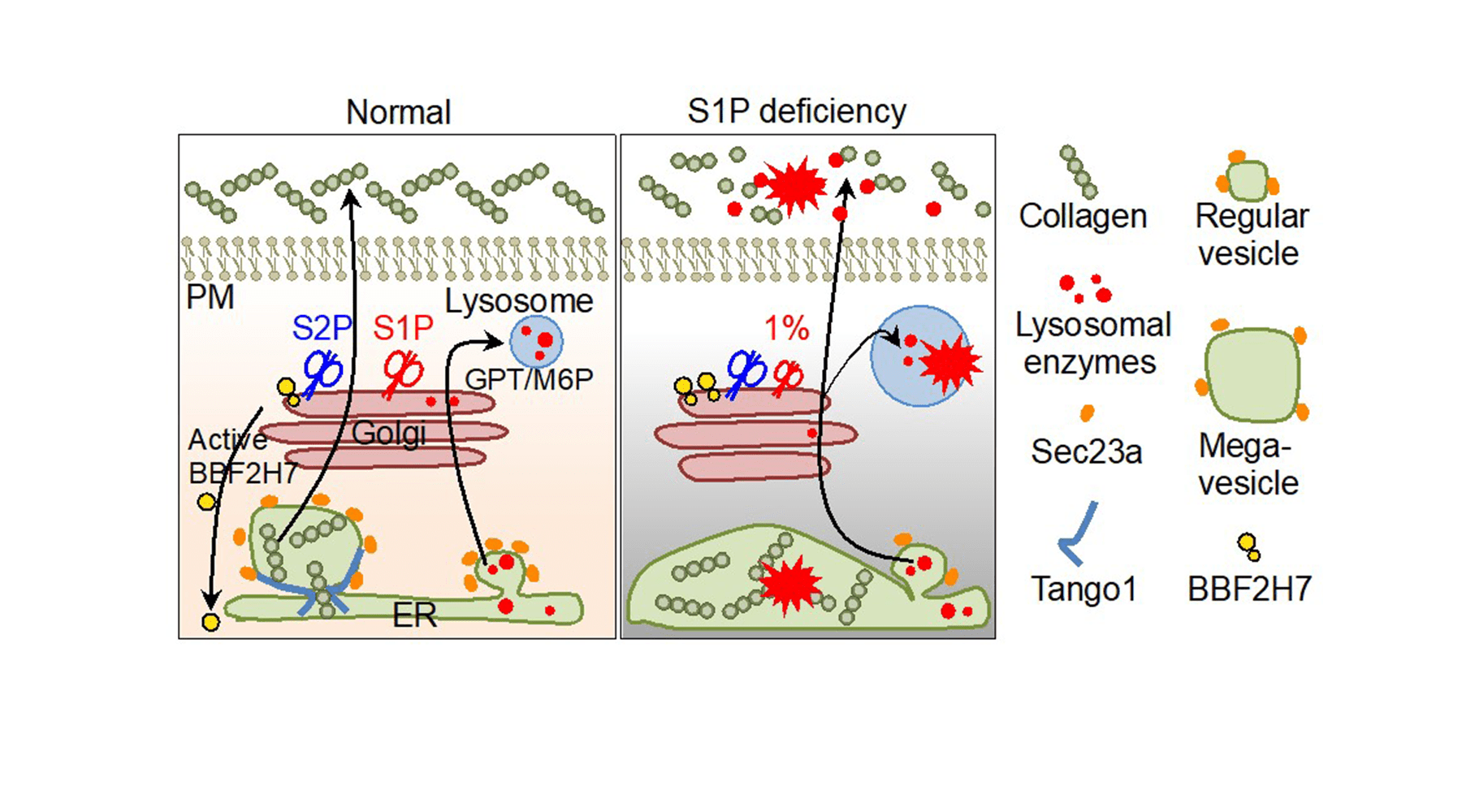
MBTPS1 (membrane bound transcription factor peptidase, site 1) encodes a protease named site-1 protease (S1P) in the Golgi. S1P is considered a master regulator of lipid homeostasis, endoplasmic reticulum (ER) stress responses, and lysosome biogenesis in mice or in cultured cells. However, how S1P differentially regulates these diverse functions in humans has been unclear.
In 2018, we discovered a pediatric patient with severe hypomorphic MBTPS1 composite mutations, resulting in severely retarded growth and skeletal anomalies, including spondyloepiphyseal dysplasia with associated kyphosis and reduced bone mineral density. Elevated levels of blood lysosomal enzymes and congenital cataracts have also been observed. The disease was named after the first two authors of our research group as spondyloepiphyseal dysplasia, Kondo-Fu type (SEDKF).
MBTPS1 is essential for the formation of COP-II vesicle, which is critical for transport of nascent proteins between ER and Golgi. Uniquely, COP-II vesicles must be enlarged to accommodate huge molecules such as collagen. This mechanism involves several unique proteins. Of such proteins, heat shock protein 47 (Hsp47), a collagen-specific ER chaperone, binds to collagen for protein folding. Sedlin is recruited to ER membrane and facilitates the growth of COP-II vesicle. Tango1 is ER-localized transmembrane protein and required for accommodation of collagen into mega-vesicle through direct binding to collagen.
MBTPS1-related disorders are a group of genetic diseases that often have secretion defects in chondrocytes in the cartilages that cause skeletal dysplasia together and the following growth retardation due to genetic mutations of key regulators as described above in the secretory machinery. If too much collagen or other cargos fail to be secreted from cells and eventually accumulates within the cells, that can cause the skeletal organs to malfunction. Men and women of all races and ethnicities can be affected.
We aim to identify more patients so that we can understand the cause of these disorders. For more information, please go to https://www.omim.org/entry/618392; or https://insight.jci.org/articles/view/121596


